Research
Cancer is a genetic disease and is fueled by the accumulation of genomic alterations that improve the fitness of the cell. A tumor cell is exposed to a plethora of intrinsic and extrinsic stimuli that can act to stimulate or inhibit the progression of the disease. How a tumor cell escapes and evolves in time and space to cause treatment-resistance and lethal disease is a fundamental question in cancer research. We are interested in the mutational mechanisms during tumor evolution (Figure 1), in particular, the mechanisms and functional consequences of large-scale genomic structural variations (SV) (e.g. deletions, duplications, and translocations) in cancer.
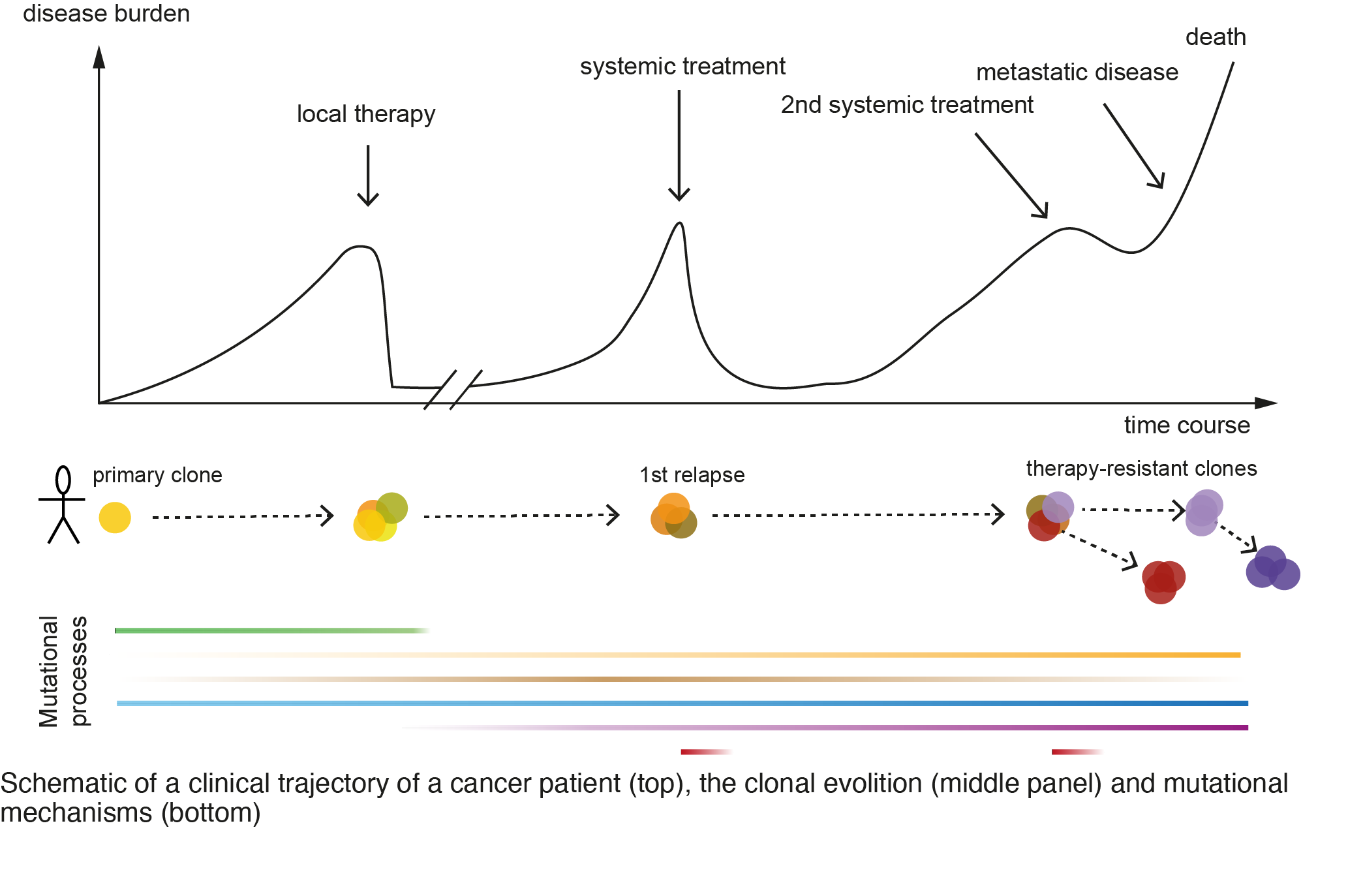
The formation of SVs is influenced by inherited, acquired, and environmental factors that together shape the (epi-)genomic architecture. We are using computational classification methods to detect and infer patterns of rearrangements (Gerhaüser et al, Cancer Cell, 2018; Rheinbay et al, Nature, 2020; Li et al, Nature, 2020), to study genotype-phenotype associations in cancer and to infer drug-sensitivity from mutational signatures. We found an age-dependent hormone-driven pathomechanism, which led to gross differences in the formation and type of SVs in early versus late-onset cancer patients (Weischenfeldt et al. Cancer Cell, 2013) and we are studying the interplay between mutational mechanisms and disease progression in this context (Figure 1). We develop and apply mathematical models to study how tumors evolve and to predict how mutations can affect the clinical trajectories of the cancer cell. To this end, we have developed PRESCIENT (Gerhaüser et al, Cancer Cell, 2018), a conditional probabilistic framework to predict the molecular changes and associated outcomes in a clinical context.
We are also using single-cell based methodologies to gain further insight into the clonal evolution and cell-type compositions that influence the tumor cells. A recurrent observation we made was the importance of chromatin organization and the epigenetic landscape in determining where breakpoints occur (Gerhaüser et al, Cancer Cell, 2018; Rheinbay et al, Nature, 2020). Most SVs occur outside of the protein-coding regions of the genome, but we found that SVs can have a drastic consequence on the 3D chromatin conformation (Figure 2) and that this can directly impact nearby gene expression, for example, by disrupting or altering hard-wired gene regulatory networks (Weischenfeldt et al, Nat Rev Genet, 2013). We developed a computational method termed CESAM (cis-expression structural alteration mapping) to identify the functional consequences of SVs on nearby gene expression. Using this method, we have performed analyses across multiple tumor types which led us to further uncover mechanisms by which distant lineage-specific enhancers get translocated and positioned in front of normally silenced oncogenes, leading to their high-level upregulation, a mechanism termed Enhancer Hijacking (Figure 2; Weischenfeldt et al, Nat Genet, 2017; Northcott et al, Nature, 2017; Rheinbay et al, Nature, 2020). Our ongoing analyses using chromatin conformation capture methodologies and computational approaches are uncovering novel and unexpected mechanisms by which alterations in the 3D chromatin organization impact gene regulation.
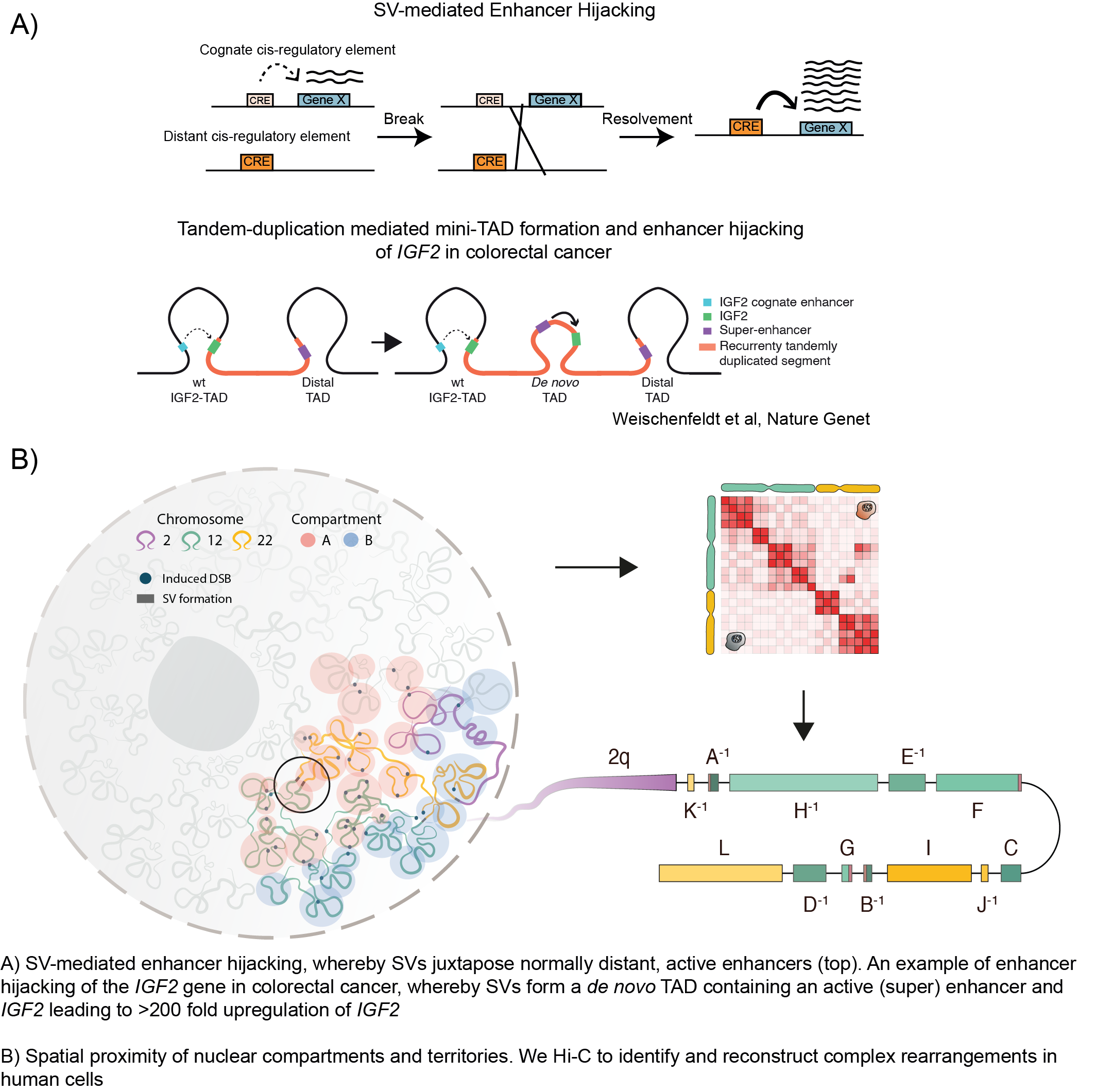
Our aim is to both improve the basic understanding of how changes in the genomic architecture impact on gene (de)regulation and to use this knowledge to prospectively identify specific, targeted therapeutic options based on the mutational profile of the tumor. To allow such inferences, we combine data generation from primary tumor samples and model systems together with integrative data analysis. Current and future research
Main projects
Clonal evolution analysis to identify recurrent mechanisms of treatment resistance and identify novel therapeutic targets
• Develop and apply methods to reconstruct the clonal evolution and to identify clinical trajectories
• Utilizing the clonal reconstruction to Identify therapeutic targets
• Pursue analysis at the single-cell level to identify clonal dynamics and interplay with the tumor microenvironment
How complex structural variants emerge and affect the 3D chromatin architecture in cancer
• Integrate epigenomic data with somatic alterations and patient-related data using data-driven quantitative genetics-based computational approaches
• Apply chromatin architecture methodologies such as Hi-C to identify how genomic alterations can impact gene regulation through altered chromatin looping
• Identify prognostic and/or predictive signatures of complex SVs
Technology
We use a combination of computational and molecular biology technologies to reach our research aims. We have optimized and integrated a range of different sequencing approaches on clinical material including paired-end sequencing, RNA-seq, ATAC-seq, Hi-C, Capture-Hi-C, mate-pair seq, DNA panel seq, single-cell sequencing, long-read sequencing. We carry out our compute on HPC systems, primarily computerome.dk. We use git for code version control and Docker to deploy our code on other systems.
We developed svscape to investigate SVs from cancer patients and associations with A and B compartments
Sustainability
Our group is committed to conducting excellent research while at the same tile reducing our environmental footprint. We implement evidence-based actions and build habits that last beyond individual team members, taking a practical, continuous-improvement approach.
Laboratory sustainability: LEAF accreditation
We participate in the Laboratory Efficiency Assessment Framework (LEAF), a structured laboratory sustainability program developed to improve the sustainability and efficiency of laboratories through a set of practical actions (e.g., reducing energy and water use, cutting waste, and improving resource management). 1
Our LEAF milestones
-
Bronze certificate — May 2024
-
Silver certificate — December 2025

LEAF helps us embed good practices into day-to-day lab work and provides a transparent framework to track progress over time.
Learn more: LEAF – Laboratory Efficiency Assessment Framework
Sustainable computing: Green DiSC enrolment
Alongside our wet-lab efforts, we are expanding our sustainability work to cover the environmental impacts of computational research. We are now enrolling in Green DiSC (Digital Sustainability Certification).
Green DiSC is an open-access certification scheme that provides a roadmap for research groups and institutions to tackle and reduce the environmental impacts of their computing activities. It is designed for teams running computation across any research domain and offers tiered certification levels (Bronze, Silver, Gold), with criteria selected to be evidence-based and practical to implement. 2
By joining Green DiSC, we aim to strengthen and formalize sustainable practices in our computational lab, including (for example):
-
choosing more energy- and carbon-efficient computing approaches when scientifically appropriate
-
improving software efficiency and computational workflows
-
reducing unnecessary storage and data transfer
-
promoting reuse, reproducibility, and responsible use of compute resources
Learn more: Green DiSC – a Digital Sustainability Certification
Governance and continuity
We have nominated Alessio Locallo and Jakob Schmidt Jespersen as the LEAF representatives and Alessio Locallo as the Green DiSC Representative for the Weischenfeldt Lab. Regarding Green DiSC, the representative is responsible for leading and coordinating the group’s sustainability efforts, as well as overseeing Green DiSC submissions and follow-up actions.
Responsibilities include:
• coordinating sustainability initiatives and tracking progress against targets
• maintaining group guidance and records relevant to Green DiSC
• acting as the contact person for sustainability-related enquiries and collaboration
Contact: alessio.locallo@bric.ku.dk
To ensure continuity, the Green DiSC Representative maintains up-to-date documentation in the group’s shared storage and will hand over responsibilities to a successor if they leave the group.